
What is Emanuel Syndrome?
A syndrome is typically defined as a set of findings that characterize a condition. Our children are usually diagnosed, however, after a positive genetic test indicating the presence of an extra small “derivative” chromosome made up of parts of chromosomes 11 & 22. Having this extra chromosome leads to the common features that are typically seen in the majority of children with ES, and which may include a small jaw (micrognathia), cleft palate, heart defects, ear differences, skin tags, kidney and genital abnormalities, and intellectual and physical impairments. It is important to know that not all children with Emanuel syndrome have exactly the same features. Some children may have fewer physical or medical issues, but all will experience delays in their intellectual development.

Our children have been said by some parents to resemble each other because of characteristic facial features, which although can vary, may include a broad nose, a longer upper lip (philtrum), low set ears, smaller chin, and deep-set eyes.
What causes Emanuel Syndrome?
Emanuel Syndrome is caused by the presence of an extra derivative chromosome, which is made up of the top part of chromosome 22 and the bottom part of chromosome 11. Typical individuals have 46 chromosomes, but individuals with Emanuel Syndrome inherit this extra derivative chromosome, which gives them 47 chromosomes in total – one too many. This is much like how individuals with Down Syndrome have an extra chromosome 21 – a trisomy 21. A trisomy is three copies of a whole chromosome or part of a chromosome.
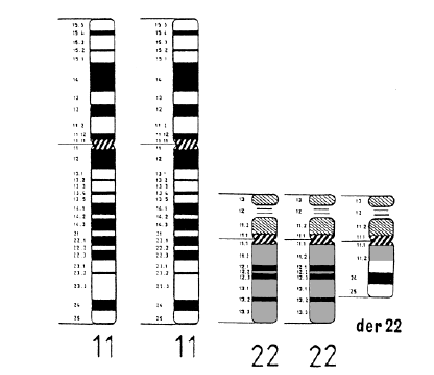
Chromosomes come in pairs, and these pairs are numbered from 1 to 22 along with a pair of sex chromosomes – either two X chromosomes (for a female) or an X and a Y chromosome (for a male). When a baby is conceived, he or she inherits one set of chromosomes from each parent. In the case of 11/22 balanced translocation carriers, to produce a child who has Emanuel syndrome, either their egg or sperm would have had to contain the derivative chromosome.
Thus, in ES, a part of chromosome 22 is present three times instead of the usual two times, from the very top, or “p” arm, down to the long “q” arm, to the breakpoint identified as 22q11.2. Part of chromosome 11 is also present three times instead of the usual two times, from the 11q23.3 section down to the end.
Having too much or too little genetic material (such as a missing or extra part of a chromosome) very often results in birth defects. Since balanced carriers have the correct amount of genetic material that has simply switched places, they develop normally. If the chromosomes are the blueprint for how cells form, problems with the chromosomes are like faulty blueprints that cause cells (and therefore the tissues and organs that cells make up, such as the brain and heart) to develop abnormally.
Individuals with Emanuel Syndrome will have a karyotype that reads:
47, XX (or XY), +der(22),t(11;22)(q23.3q11.2), mat (or pat)
A karyotype is basically a word map that describes a person’s chromosomes.
47 – indicates that there are 47 chromosomes (a normal complement is 46)
XX – indicates if it is a female (XY if it is a male)
+der(22) – indicates that there is an extra (+) “derivative chromosome” (der) present, and the number in the brackets indicates which chromosome is involved (based on the centromere, which holds the p and q arms together – NOT on the larger piece of chromosome)
t(11;22) – refers to the two chromosomes involved in the translocation
(q23.3;q11.2) – refers to the breakpoints involved on each chromosome
The karyotype will be followed by “mat” if the extra chromosome was inherited from the mother (maternal) or “pat” (paternal) if it was inherited from the father. In rare instances, the child may also inherit the balanced translocation as well as the extra chromosome.
Karyotypes that were done before the mid-1990s may show different breakpoints on these 2 chromosomes, but Dr. Emanuel’s research has since shown that the breakpoints are almost always the same, so older reported karyotypes that differ are presumed to be inaccurate now. They were done with the best technology available at the time.
Information on Genetic Testing from National Library of Medicine
Dr. Emanuel’s original article from one of our group’s first newsletters in 1996 was written before we had the condition named. The article in its original format:
By Dr. Beverly Emanuel,
The Children’s Hospital of Philadelphia
In order to talk about the Supernumerary der(22) Syndrome, first we need to step back and describe some terminology. This is so that everyone reading this will begin on the same footing. As you may know, a syndrome is really a collection of findings that has been seen recurring over and over again in patients. For example, one group of associated features actually includes: a heart problem; malformed ears with pits or tags; small chin; and a high arched or cleft palate. Syndromes are often named after the person or persons who first described the collection of findings, although this has not happened in this case. Once an underlying cause is identified, the name may be changed to reflect the specific chemical abnormality, chromosome difference, or gene change that caused the problem. Here, in the case of the Supernumerary der(22)t(11;22), the name reflects the chromosomal change.

Genes are made up of a chemical called DNA and they are housed within larger structures called chromosomes. Most people have 23 pairs of chromosomes (46 total), with one of each pair coming from the mother and the other from the father. Chromosomes are numbered 1 through 22; the 23rd pair are called sex chromosomes because they determine a person’s sex (male or female). The chromosomes are found in every cell in the body. Cells are so small that they, and the chromosomes they contain, can only be seen by observation with a microscope.
Since genes are housed inside the chromosomes, they themselves can’t be seen at the microscope, but they can be measured by using special “molecular” tests. A good way to think about chromosomes and genes is to compare them to a train. A train has a number of box cars just as a chromosome has a number of stripes or bands. We can see the box cars when we look at a train, just as we can see the chromosomes and their band patterns when we observe them at the microscope. We cannot, however, see the packages inside the box car without first opening the door. The same is true for a chromosome – the genes are the packages inside.
When a baby is conceived with either too much or too little chromosomal material, birth problems or birth defects can occur. This may include a whole extra chromosome, as in the Supernumerary der(22)t(11;22) syndrome (an extra “derivative” 22 chromosome), a whole missing chromosome as in Turner syndrome (a missing X), a piece of material missing or extra, or a complex rearrangement of chromosomal material. When chromosomal material is missing or extra, genes are generally missing or extra. Since genes are the blueprint of the body, when they are deficient or duplicated, the body’s blueprint changes, frequently leading to birth problems and learning differences.
So again you ask, what is the 11;22 translocation and the Supernumerary der(22) Syndrome?
In 1980, working at the Children’s Hospital of Philadelphia in the U.S.A., we (Dr. Elaine Zackai and I) described the 11;22 translocation. At about the same time, the t(11;22) was also described by a consortium of European scientists. People who carry the 11;22 translocation have a very small piece of chromosome 22 (22q11 -> qter) transferred to chromosome 11 and a small piece of chromosome 11 (11q23 -> qter) transferred to 22 (thus, it is called a translocation). Chromosomes are divided into two parts, the top part being called the “p”or short arm and the bottom part called the “q” or long arm. Thus, the 22q11 -> qter and 11q23 -> qter designation tells everyone who works in genetics that the area transferred or translocated starts at a very specific spot on the “q” arm of chromosomes 11 and 22 and goes to the end (“ter” or terminus) of the “q” arm. It is very important to know the location of a moved piece of chromosomal material in order to make some general comparisons between individuals. This is because if two children have different parts of the same chromosomes extra it would be like comparing “apples to oranges” to compare them to one another. Most often when there is a chromosomal rearrangement or translocation they are not exactly alike.
However, with the t(11;22), we suspect that the story is different. The 11;22 translocation is appears to be the only translocation which seems to have recurred over and over again, creating numerous carriers. The points of chromosome exchange appear to be at the same spots on 11 and 22 in all instances. Several hundred families with what appears to be the same rearrangement have been described in the scientific literature. Carriers of the t(11;22) are themselves normal, but come to the attention of the geneticist or pediatrician subsequent to the birth of a child affected with the +der(22) syndrome who has the derivative (22) or rearranged chromosome 22 as an extra chromosome. Occasionally, translocation carriers are discovered upon being studied for multiple miscarriages or infertility. Patients with the +der(22) syndrome (or the extra chromosome) have distinctive features which can include tags or pits in front of their ears, abnormally shaped ears, a cleft or high arched palate, a small chin, a heart defect, mental retardation and sometimes genital abnormalities in the male.
Geneticists have long been interested in understanding the mechanisms and results of chromosomal rearrangement. Chromosomal rearrangements or translocations are the result of DNA rearrangements at the molecular level. Translocations are one of the major categories of structural chromosomal alterations. The mechanisms by which they are generated are largely unknown. Balanced translocations, i.e. those in which there is no microscopically visible loss of genetic material are amongst the most common of these rearrangements accounting for roughly 0.2% of structural abnormalities. In general, the chromosomal change is presumed to occur during the formation of either the egg or sperm which created the first person with the rearrangement in a family. Then, that person carries the change in every cell of his or her body and is capable of passing the change to his/her children. It can be transmitted either as a balanced rearrangement or as an extra chromosome as in the Supernumerary der(22) Syndrome.
The cellular events which end in chromosomal rearrangement or translocations, especially those like the recurrent t(11;22), remain to be elucidated. Understanding the molecular basis of this translocation may serve as a model for understanding the mechanisms involved in creating other translocations. Thus, efforts to map the elusive constitutional t(11;22) translocation breakpoint are progressing. For example, we have already determined that a major portion of the DNA which surrounds the region of the breakpoint on chromosome 22 is duplicated several times on chromosome 22. This duplication which is normally present on every chromosome 22 may make this chromosome more prone to engage in rearrangements. The determination of the exact size of the duplication, number of copies and their location is currently under investigation. Further, the relationship between having a translocation, especially an 11;22 translocation, and how chromosomes separate during formation of egg and sperm, is not clear and has not been well studied. These are some of the studies we are currently pursuing in our laboratory research.
Further, our research team includes several geneticists who specialize in identifying genes, how they work, and why they cause problems when they are extra or changed. We hope to provide the scientific community with new information about the genes which reside in the duplicated pieces of chromosomes 11 and 22. We also try to understand how genes influence each other. When there is a supernumerary der(22), many genes are extra but probably not all of them play a role in causing the symptoms associated with the duplication. Thus, additional studies will be necessary to understand the role that each of the genes plays in the cause of the complex and variable symptoms which are seen in the children with the Supernumerary der(22) syndrome. Our research studies will enable us to determine if these problems are caused by the duplication of several genes, or by many duplicated genes. Further, most individuals with the Supernumerary der(22) syndrome have a duplication of the same exact pieces of chromosomal material but they do not all have the same symptoms. Perhaps other factors play a role in who develops which symptoms. Much more work needs to be done and this is our job for the future. We welcome your participation in our research efforts and thank you for all of your interest. TOGETHER, we will begin to understand the t(11;22) and the Supernumerary der(22)t(11;22) syndrome.